This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
Amytrophic Lateral Sclerosis and FUS [1,2,3]
|
Amyotrophic lateral sclerosis (ALS) is a neurological disease that involves the degeneration and death of motor neurons necessary for producing voluntary muscle movement. Symptoms of ALS generally include spasticity and muscle weakness (Figure 1). As the disease progresses to upper motor neurons, individuals suffer cognitive impairments and respiratory insufficiency (Figure 1). The Centers for Disease Control and Prevention estimates that roughly 14,000-15,000 Americans were living with ALS in 2016. ALS predominately affects males ages 55-75, but can appear in all races, demographics, and ages. In approximately 5-10% of cases, it is shown that ALS originates primarily through genetic heritage. Most of these cases are associated with various pathogenic variants in C9orf72, TDP-43, and FUS.
As of today, at least 85 mutations have been associated with the FUS gene and amyotrophic lateral sclerosis (ALS). Specifically, for FUS gene alterations, these cases tend to develop a more aggressive juvenile-onset form of ALS occuring around 20 years of age. This suggests some important disrupted cellular mechanism is causing early death in these individuals. |

Figure 1: Common Symptoms of ALS
|
FUS: RNA/DNA Binding Protein [4,5,6]

Figure 2: Known Human FUS Pathogenic Variants
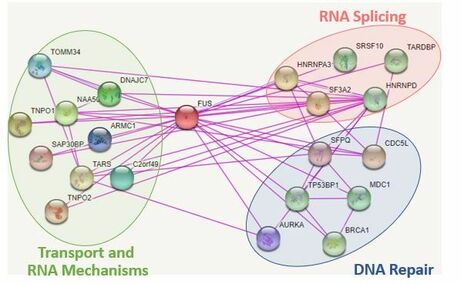
Figure 3: Known Human FUS Interactors
|
FUS has been shown to contain two main domains, the RNA recognition motif and zinc finger domain. These regions are known to play critical roles in DNA and RNA binding. The mutations in these diseased states tend to localize in two main regions near the C terminal region and the RNA recognition motif (Figure 2). These genetic changes are believed to alter the DNA binding and mRNA binding ability of the protein. FUS is normally localized in the nucleus, but in diseased states the protein becomes trapped in the cytoplasm. When FUS is localized to the nucleus, it has been shown to play biological roles in RNA splicing, metabolism, and most interestingly, DNA repair (Figure 3). Recently, the importance of FUS in DNA repair is beginning to gain more attention. Part of this reason results from the similarities of DNA damage shown in other neurodegenerative diseases. Due to this similarity and juvenile onset of FUS, I chose to focus my project on understanding how DNA repair defects lead to neurodegeneration.
|
Gap in Knowledge [7,8,9]
|
Individuals with FUS mutations have been shown to possess elevated levels of single strand and double strand breaks. Protein interactions of FUS in DNA repair have been suggested, but the exact mechanism of FUS in single strand and double strand break repair is not well understood (Figure 4). It has been shown that altered FUS in DNA repair facilitates aggregate formation, which directly lead to neurodegenerative disease.
Although it is known that impaired DNA damage response mechanisms help facilitate FUS aggregates, it is unknown how defects in the DNA repair pathways lead to neuron defects. By understanding the mechanisms of FUS involved in DNA repair mechanisms in both single strand and double strand breaks, potential therapeutic targets could be identified to prevent aggregate formation and neurodegeneration. |

Figure 4: Proposed Base Excision Repair Mechanism
|
Project Aim and Hypothesis
The objective of this study is to determine how FUS-related DNA repair leads to proper motor neuron function. I hypothesize that FUS domains play roles in DNA repair mechanisms, due to their conserved nature and role in DNA binding. The long-term goal of this research is to determine how FUS-related DNA repair leads to neurodegeneration.
Model Organism
Danio rerio would be a useful model for studying alterations to the FUS gene and analyzing the association with both ALS and DNA repair defects [10]. These organisms contain multiple conserved pathogenic variant sites, possess both conserved domains of FUS (Figure 5), and share a conserved evolutionary history (Figure 6). In addition, these organisms are relatively cheap, easy to maintain, and possess transparent embryos for analyzing motor neuron defects. The phenotypes of motor movement and use of a comet assay can provide an efficient way of analyzing motor neuron degeneration and DNA damage (Figure 7,8). By using this model organism, it would be relatively efficient to screen for DNA repair defects associated with FUS and symptoms of ALS.

Figure 5: Conserved Domains of FUS
|
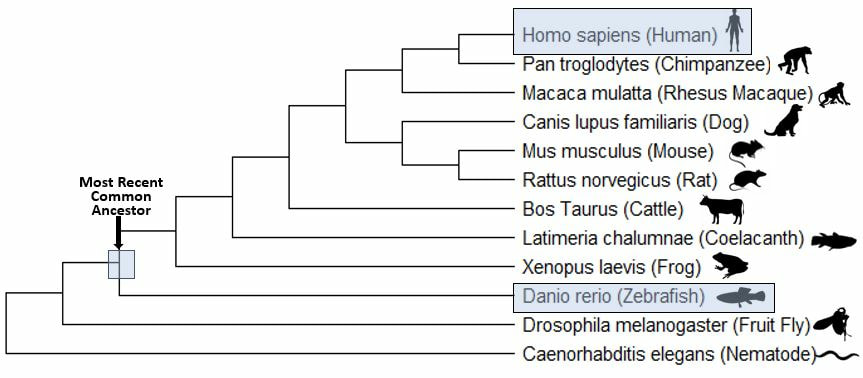
Figure 6: Evolutionary History of FUS
|
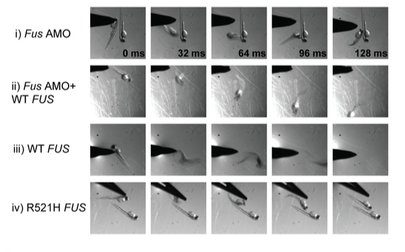
Figure 7: Motor Movement Screen of FUS
|
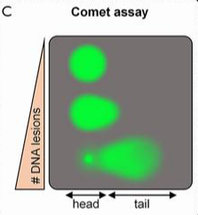
Figure 8: Comet Assay Visualization
|
Specific Aims
Specific Aim 1: Identify amino acids of FUS in DNA repair and neurodegeneration
I will perform a protein alignment to generate CRISPR mutants in order to find conserved regions associated with DNA repair in FUS (Figure 9). To begin, I will align protein sequences via MEGA to identify conserved amino acids in the RNA recognition motif, zinc-finger domain, and those that contain conserved pathogenic variants, like G187C [4]. As controls, I will use wild type and a R521C mutant, already shown to have high DNA damage [11]. I will then utilize CRISPR-Cas9 technology to induce specific mutations along those conserved regions. Sanger sequencing will then be used to confirm that the correct alterations were made. Next, I will screen for phenotypes showing defective motor movements. For those showing motor defects, I will measure the amount of single/double stranded breaks (SSBs/DSBs) via a comet assay when subjected to UV damage [11] (Figure 8). This method will allow for a quantification of DNA damage present in each neuronal sample and highlight regions associated with DNA repair.
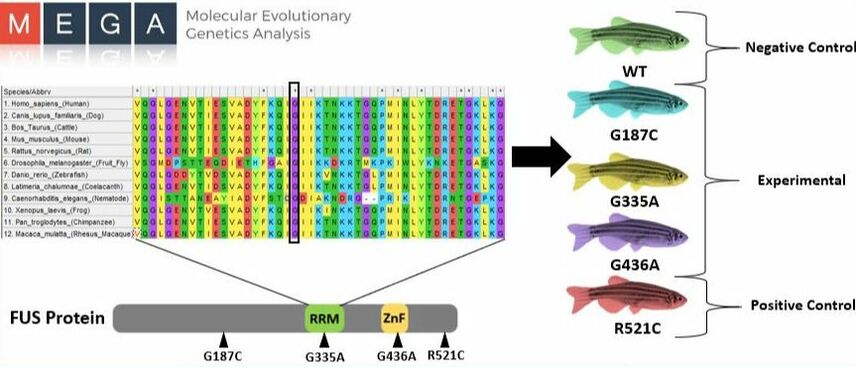
Figure 9: Alignment of Critical FUS sites
I hypothesize that FUS mutations in the zinc finger domain will impact DNA repair and result in more SSBs/DSBs, due to zinc finger domains emerging roles in genome stability [12]. Screening of D. rerio with these specific variants should result in a phenotype with increased SSBs/DSBs, which would define DNA repair binding regions important for genome stability and neuronal function. This knowledge would provide us with more insight into the mechanism of FUS in DNA repair and potential therapeutic targets for those containing the specific variants.
Specific Aim 2: Identify small molecules that rescue FUS mutant phenotype
I will perform a chemical screen by using a known CRISPR-Cas9 mutant, R521C, that has showed increased SSBs/DSBs and motor movement impairments [11] (Figure 10). To begin, I will utilize a chemical library of small molecules that regulate DNA repair proteins involved with SSBs/DSBs, like those in homology directed repair and base excision repair [14]. Specifically, it has been suggested that a PARG inhibitor could help promote DNA repair for the base excision repair pathway. For my chemical library, I would test multiple FDA approved drugs that could alter specific proteins in DNA repair pathways. After the developing Danio rerio are subjected to these protein regulators, I will perform a motor movement screen and comet assay to confirm if any were restored to a wild type phenotype.
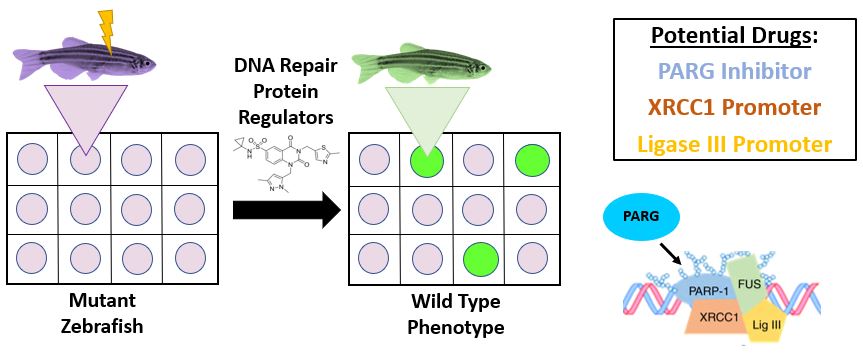
Figure 10: Overview of Chemical Screen
I hypothesize that small molecules associated with the down-regulation of proteins that terminate repair complexes will rescue the FUS mutant phenotype, due to increased retention time at SSBs/DSBs sites. By identifying small molecules that restore DNA repair and motor neuron function, FDA approved cancer drugs could easily be transitioned to ALS patients. Currently, there are no drugs that have been shown to effectively prolong lifespan or reverse the effects of the disease. Thus, showing the importance of conducting a chemical screen to identify new drug options.
Specific Aim 3: Identify new FUS protein-protein interactions in DNA repair
Utilizing lysed neuronal cells from D. rerio, I will subject samples to both wild type and mutant FUS baits (R521C). Tandem affinity purification and mass spectrometry will then be used to purify and obtain MS/MS data (Figure 11). Computer software would provide sequence coverage of these captured protein interactions. To confirm these interactions are legitimate, CRISPR-Cas9 will be used to make knockouts of potential FUS binding sites determined by BLAST (Figure 12). A comet assay and motor movement screen will identify DNA repair proteins as those with increased SSBs/DSBs and possible motor neuron defects. Proteins will then be sorted by GO terms, which would elucidate a more extensive protein interaction network of FUS in DNA repair.

Figure 11: Tandem Affinity Purification and Mass Spectrometry
|
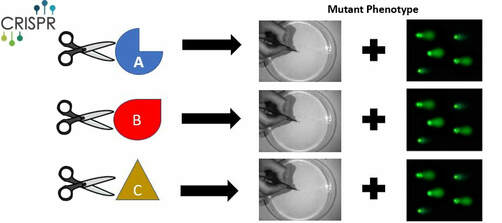
Figure 12: CRISPR/Cas9 Knockout of New Protein Interactors
|
I hypothesize that TAP-MS will provide new FUS interactions associated with DNA repair in D. rerio, due to the presence of high levels of SSBs/DSBs commonly found in FUS mutants. Currently, databases like String show limited DNA repair protein interactions with FUS in D. rerio [15]. Eukaryotic DNA repair pathways are also well conserved, and this data could suggest similar mechanisms present in humans [16]. This approach would expanded the FUS interaction network and provide additional therapeutic targets that could be altered in diseased states.
Future Directions/Conclusion
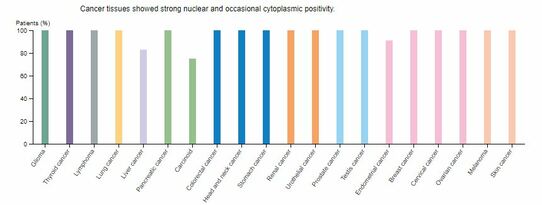
Figure 13: Expression of FUS in Cancer
|
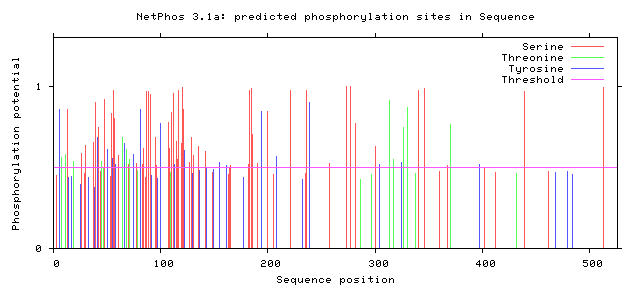
Figure 14: Phosphorylation Sites of Human FUS
|
Multiple studies fail to address how FUS-ALS individuals fail to develop cancer with elevated DNA damage. In one study, it was shown that high levels of FUS tend to have negative outcomes in lung cancer. FUS is also highly expressed in numerous cancer types, yet cancer rarely develops in FUS-ALS individuals (Figure 13). A future goal would be to utilize RNA-seq or quantitative proteomics to examine expression of RNA and proteins. The main idea is that cell cycle checkpoint inhibitors could be upregulated in these patients. It is possible that this DNA damage upregulates those processes and could explain why we see an earlier death in these individuals. A second direction would be to look at drug administration at different time points. It is unclear if drug administration can restore motor neuron function in older organisms. It is expected that DNA damage defects will be restored, but whether motor neuron function can be restored after prolonged damage is up for discussion. A third area to research would be to look at phosphorylation sites that correspond to human variants (Figure 14). I chose not to focus on phosphorylation sites, since most failed to align with all human variants and show conservation across species. However, there were two human variants that corresponded to potential phosphorylation sites. Considering the role of phosphorylation in DNA repair pathways, it would be important to look more into these two sites.
Through these approaches, the role that FUS plays in DNA repair and motor neuron function can be identified. As of today, there are no known therapeutic treatments for FUS-ALS patients that can reverse motor neuron defects. By understanding targets of FUS in the DNA repair pathways, current FDA approved cancer drugs targeting such pathways could easily be administered to those with FUS-ALS. This would not only help restore genomic stability, but also provide the potential for restored neuron function in patients. Ultimately, this understanding would highlight how DNA repair defects lead to neurodegeneration.
Through these approaches, the role that FUS plays in DNA repair and motor neuron function can be identified. As of today, there are no known therapeutic treatments for FUS-ALS patients that can reverse motor neuron defects. By understanding targets of FUS in the DNA repair pathways, current FDA approved cancer drugs targeting such pathways could easily be administered to those with FUS-ALS. This would not only help restore genomic stability, but also provide the potential for restored neuron function in patients. Ultimately, this understanding would highlight how DNA repair defects lead to neurodegeneration.
First Draft Presentation: |
Final Presentation:
| ||
References:
[1] Amyotrophic Lateral Sclerosis (ALS) Fact Sheet. (2018, August 9). Retrieved February 3, 2019 from https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet
[2] Amyotrophic lateral sclerosis (ALS). (2018, July 17). Retrieved February 3, 2019 from https://www.mayoclinic.org/diseases-conditions/amyotrophic-lateral-sclerosis/symptoms-causes/syc-2035402
[3] Zou, Z.Y., Liu, M.S., Li, X.G., Cui, L.Y. (2015, September). Mutations in SOD1 and FUS caused juvenile-onset sporadic amyotrophic lateral sclerosis with aggressive progression. Ann Translation Medicine 3(15):221
[4] Shang, Y. & Huang E.J. (2016, September). Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Research 1647:65-78.
[5] Conte, A., Lattante, S., et al. (2012, January). P525L FUS mutation is consistently associated with a severe form of juvenile Amyotrophic Lateral Sclerosis. Neurology Genetics 2:6
[6] Zhou, Y., Liu, S., et al. (2013, October). ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. Nature Communications 9:368
[7] Wang, H., Guo, W., et. Al. (2018, September). Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nature Communications 9:3683
[8] Naumann, M., Pal, A., et al. (2018, January). Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nature Communications 9:335
[9] Penndorf, D., Witte, O., et al. (2018, February). DNA plasticity and damage in amyotrophic lateral sclerosis. Neural Regeneration Research 3(2): 173–180.
[10] McGown, A., McDearmid, J.R., et al. (2012, October). Early interneuron dysfunction in ALS: Insights from a mutant sod1 zebrafish. Annals of Neurobiology 73(2):246-258.
[11] Qiu H., Lee S., Shang Y., Wang WY., et al. (2014, March). ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest 124(3):981-99
[12] Vilas C.K., Emery L.E., et. al. (2018, January). Caught with One’s Zinc Fingers in the Genomce Integrity Cookie Jar. Trends Genetics 34(4):313-325.
[13] Naumann M, Pal A, Goswami A, Lojewski X, et. al. (2018, January). Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nature Communications 9(1):335.
[14] Dance, A. Targeting FUS: DNA Damage Control in ALS. The ALS Research Form. Retrieved on May 2, 2019 from https://www.alsresearchforum.org/targeting-fus-dna-damage-control-in-als/
[15] SMART. Retrieved on 4/11/2019 from https://version11.string-db.org/cgi/input.pl?sessionId=Odz7uTVCbz7d&input_page_show_search=on
[16] Taylor E.M., Lehmann, A.R. (1998, September). Conservation of eukayotic DNA repair mechanisms. Int J Radiat Biol 74(3):277-86.
[2] Amyotrophic lateral sclerosis (ALS). (2018, July 17). Retrieved February 3, 2019 from https://www.mayoclinic.org/diseases-conditions/amyotrophic-lateral-sclerosis/symptoms-causes/syc-2035402
[3] Zou, Z.Y., Liu, M.S., Li, X.G., Cui, L.Y. (2015, September). Mutations in SOD1 and FUS caused juvenile-onset sporadic amyotrophic lateral sclerosis with aggressive progression. Ann Translation Medicine 3(15):221
[4] Shang, Y. & Huang E.J. (2016, September). Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Research 1647:65-78.
[5] Conte, A., Lattante, S., et al. (2012, January). P525L FUS mutation is consistently associated with a severe form of juvenile Amyotrophic Lateral Sclerosis. Neurology Genetics 2:6
[6] Zhou, Y., Liu, S., et al. (2013, October). ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. Nature Communications 9:368
[7] Wang, H., Guo, W., et. Al. (2018, September). Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nature Communications 9:3683
[8] Naumann, M., Pal, A., et al. (2018, January). Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nature Communications 9:335
[9] Penndorf, D., Witte, O., et al. (2018, February). DNA plasticity and damage in amyotrophic lateral sclerosis. Neural Regeneration Research 3(2): 173–180.
[10] McGown, A., McDearmid, J.R., et al. (2012, October). Early interneuron dysfunction in ALS: Insights from a mutant sod1 zebrafish. Annals of Neurobiology 73(2):246-258.
[11] Qiu H., Lee S., Shang Y., Wang WY., et al. (2014, March). ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest 124(3):981-99
[12] Vilas C.K., Emery L.E., et. al. (2018, January). Caught with One’s Zinc Fingers in the Genomce Integrity Cookie Jar. Trends Genetics 34(4):313-325.
[13] Naumann M, Pal A, Goswami A, Lojewski X, et. al. (2018, January). Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nature Communications 9(1):335.
[14] Dance, A. Targeting FUS: DNA Damage Control in ALS. The ALS Research Form. Retrieved on May 2, 2019 from https://www.alsresearchforum.org/targeting-fus-dna-damage-control-in-als/
[15] SMART. Retrieved on 4/11/2019 from https://version11.string-db.org/cgi/input.pl?sessionId=Odz7uTVCbz7d&input_page_show_search=on
[16] Taylor E.M., Lehmann, A.R. (1998, September). Conservation of eukayotic DNA repair mechanisms. Int J Radiat Biol 74(3):277-86.
Header Image: https://singularityhub.com/wp-content/uploads/2018/03/neurons-brain-1-1068x601.jpg
Figures 1-14: Images are all hyperlinked to their original source or created by Nathan Johnson
Note: Powerpoint figures are also hyperlinked to their original source in the Final Presentation file.
Figures 1-14: Images are all hyperlinked to their original source or created by Nathan Johnson
Note: Powerpoint figures are also hyperlinked to their original source in the Final Presentation file.
